
Sadržaj
Cistična fibroza (CF) nasljedni je, po život opasan poremećaj koji oštećuje pluća i probavni trakt. Uzrokovan je neispravnim genom koji pokreće stvaranje zgusnute sluzi koja začepljuje dišne putove i blokira izlučivanje probavnih enzima.Simptomi su progresivni i često ozbiljni, a mogu uključivati probleme s disanjem, ponovljene infekcije pluća, loš rast, mušku neplodnost i kroničnu upalu gušterače, jetre, bubrega i srca.
CF se može dijagnosticirati krvnim testovima, genetskim pregledom i postupkom poznatim kao test znojnog klorida.
Iako ne postoji lijek za CF, postoje tretmani koji mogu poboljšati i duljinu i kvalitetu nečijeg života.
To uključuje tehnike čišćenja dišnih putova, inhalacijske antibiotike, razrjeđivače sluzi, enzime gušterače, visokokaloričnu prehranu i lijekove novije generacije poznate kao CFTR modulatori. U težim slučajevima može biti potrebna transplantacija pluća.
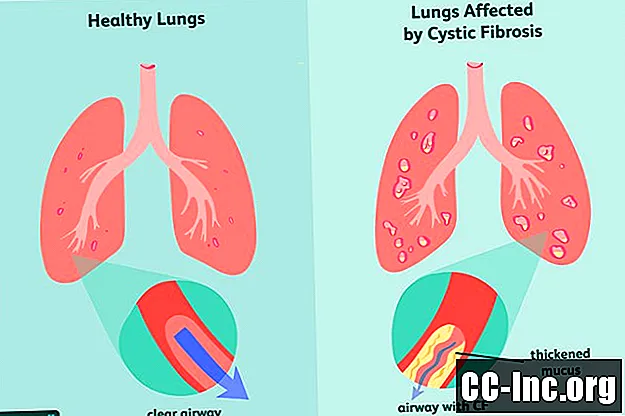
Simptomi cistične fibroze
Kao genetski poremećaj, cistična fibroza je nešto s čime ste rođeni. Može se pojaviti ili ne mora imati simptome u vrijeme rođenja, a često mogu proći mjeseci ili čak godine prije nego što se pojave znakovi bolesti. U to su vrijeme pluća i probavni trakt već imali oštećenja koja se ne mogu poništiti.
Najčešći rani znakovi i simptomi CF uključuju:
- Blokada djetetove prve stolice (mekonij)
- Koža slanog okusa
- Kronični kašalj, piskanje ili obojeni ispljuvak
- Tekuće, masne i obično smrdljive stolice
- Infekcija pluća, često se ponavlja
- Loš rast i neuspjeh u napredovanju
Ako se ovi simptomi ne mogu kontrolirati, stres na plućima (i nemogućnost debljanja) može imati kumulativni učinak, utječući na više organa i povećavajući rizik od komplikacija bolesti.
Neke od karakterističnijih komplikacija uključuju:
- Odgođeni pubertet
- Bronhiektazije (kronično zadebljanje plućnih zidova)
- Gubitak težine
- Pankreatitis (upala gušterače)
- Muška neplodnost
- Plućna hipertenzija (visoki krvni tlak u plućima)
- Žučni kamenci
- Dijabetes povezan sa cističnom fibrozom
- Cor pulmonale (zatajenje srca s desne strane)
- Ciroza (funkcionalno ožiljci jetre)
Budući da CF uzrokuje progresivne ozljede stanica i tkiva, svaka šteta nanesena plućima i drugim organima bit će uglavnom nepovratna. Smrt će najčešće biti posljedica zatajenja dišnog sustava, nakon čega slijedi zatajenje srca i zatajenje jetre.
Simptomi cistične fibroze
Uzroci
Cistična fibroza uzrokovana je mutacijom gena za transmembranski receptor cistične fibroze (CFTR), koji je odgovoran za proizvodnju proteina CFTR. To je protein koji je tijelu potreban za regulaciju protoka soli i vode u i iz stanica. . Ako je protein deformiran ili neispravan, može izazvati dehidraciju na površini stanice, što dovodi do zadebljanja okolne sluzi.
CF je autosomno recesivni poremećaj, što znači da trebate naslijediti CFTR mutaciju i od majke i od oca da biste imali bolest. Ako naslijedite samo jedan defektni gen, nećete imati CF, već ćete biti nositelj mutiranog gena.
Možete naslijediti bolest ako svaki od vaših roditelja ima CFTR mutaciju ili sam CF. Ako su oba roditelja nositelji, imali biste:
- 25 posto šanse za CF
- 50 posto šanse da budete prijevoznik
- 25 posto šanse da na to ne utječe
S druge strane, ako je jedan od vaših roditelja prijevoznik, a drugi ima CF, imate 50/50 šanse da imate CF ili da ste prijevoznik.
Cistična fibroza jedna je od najčešćih genetskih bolesti, koja pogađa otprilike jednu na svakih 2500 beba rođenih u Sjedinjenim Državama.
Najčešći je među bijelcima i Latinoamerikancima, a rjeđe se javlja kod ljudi afričkog ili azijskog podrijetla.
Čimbenici rizika od cistične fibrozeDijagnoza
Postoji nekoliko testova koji se koriste za dijagnozu cistične fibroze. Djeluju ili izravnim otkrivanjem mutacije CFTR ili neizravnim mjerenjem bioloških promjena u skladu s bolešću. Metoda dijagnoze može se razlikovati tijekom trudnoće, kada se dijete rodi ili bilo kada nakon toga.
Vodič za diskusiju liječnika cistične fibroze
Nabavite naš vodič za ispis za sljedeći pregled kod liječnika koji će vam pomoći da postavite prava pitanja.

Od dva standardna testa koja se obično koriste za dijagnozu CF:
- Ispitivanje klorida znoja, poznat i kao test znoja, mjeri količinu klorida na koži. Budući da CF ometa prijenos soli u stanice i iz njih, doći će do nakupljanja soli u znoju.
- Genetsko CFTR ispitivanje koristi se za otkrivanje najčešćih mutacija CFTR mutacije. Iako postoji preko 2000 CFTR mutacija za koje je poznato da uzrokuju cističnu fibrozu, 23 koja su uključena u standardni panel predstavljaju najizglednije osumnjičene.
Tijekom trudnoće, genetski test CFTR može se koristiti za ispitivanje tekućina dobivenih amniocentezom ili stanica ekstrahiranih uzimanjem uzoraka horionskih resica (CVS).
Probir novorođenčadi se također standardno koristi za dijagnozu CF i danas je obvezan u svih 50 država i okrugu Columbia. Što to podrazumijeva, razlikovat će se ovisno o tome gdje u Sjedinjenim Državama živite. Ako su rezultati probira za novorođenče pozitivni, test znoj će se koristiti za potvrdu dijagnoze.
Kako se dijagnosticira cistična fibrozaLiječenje
Iako ne postoji lijek za cističnu fibrozu, napredak u liječenju produžio je životni vijek onih koji žive s tom bolešću.
Cilj liječenja CF-a je četverostruk: spriječiti infekcije, zadržati plućnu funkciju, normalizirati probavu i usporiti napredovanje bolesti.
Među terapijskim alatima koji se koriste za liječenje CF:
- Tehnike čišćenja dišnih putova (ACT) izvode se za istiskivanje i izbacivanje nakupljene sluzi iz pluća. Tehnike uključuju pojačani kašalj, udaraljke u prsima ili oscilacije zida u prsima.
- Dijeta s puno masnoća i kalorija koristi se za nadoknadu malapsorpcije masti, bjelančevina i hranjivih sastojaka u crijevima.
- Dodaci enzimu gušterače koriste se za pojačavanje probavnih enzima koje gušterača ne može proizvesti zbog prekomjernog nakupljanja sluzi.
- Antibiotici uzimaju se svakodnevno radi sprečavanja bakterijskih infekcija pluća.
- Mukolitici- mogu se koristiti lijekovi koji se koriste za razrjeđivanje sluzi prije ACT-a.
- CFTR modulatori nova su klasa lijekova koji mogu ispraviti određene nedostatke u CFTR proteinu i vratiti im regulatornu funkciju.
- Terapija kisikom može se koristiti tijekom akutnih epizoda kada je vaše disanje ozbiljno oslabljeno.
- Enteralna prehrana, poznato i kao hranjenje u sondi, može se koristiti ako ne možete održati težinu normalnom prehranom.
- Transplantacija pluća smatra se kada vaša pluća više ne mogu preživjeti bez mehaničke ventilacije.
Snalaženje
1938. godine, kada je cistična fibroza prvi put klasificirana kao bolest, djeca su rijetko živjela nakon prve godine života. Do 1980-ih se moglo očekivati da će živjeti čak 20 do 25 godina. Danas se slika potpuno promijenila s ljudima koji žive dobro u svojim 40-ima, pa čak i 50-ima ako se liječenje započne rano i ako se toga pridržavaju.
To ne znači da je CF ništa manje ozbiljan nego što je ikad bio. To je događaj koji mijenja život i zahtijeva marljivost i dosljednost kako bi se ne samo suočili s bolešću već i živjeli najviši mogući životni standard.
U tu svrhu trebate normalizirati CF u svom životu uspostavljanjem rutina i praksi kako biste izbjegli uspone i padove koji mogu uzrokovati stres i povećati invaliditet. Među razmatranjima trebali biste:
- Upravljajte prehranom. Osobe s CF često trebaju dvostruko veće kalorije dnevno nego što to rade drugi ljudi.
- Redovito vježbajte. Fitness rutine idealno bi trebale uključivati najmanje 20 do 30 minuta aerobnih aktivnosti tri puta tjedno. Pronađite nešto ugodno što možete raditi čitav život.
- Držite dobro hidratiziranu. To održava pluća i crijeva ispravnim radom. Ovisno o vašoj dobi, trebali biste piti ne manje od šest do osam visokih čaša vode dnevno.
- Ispravno izvedite zračnost. Kako se vaše zdravstvene potrebe mijenjaju, tako se mogu mijenjati i vrste alata za čišćenje koje vam trebaju. Razgovarajte sa svojim pulmologom ili fizioterapeutom ako ne postižete rezultate koje biste trebali.
- Potražite podršku. Osim prijatelja i obitelji, možete kontaktirati najbliže poglavlje Zaklade za cističnu fibrozu (CFF) kako biste se uključili u mrežu podrške u vašem području.
- Potražite financijsku pomoć. CFF nudi usluge koje pomažu obiteljima da se lakše nose s visokim troškovima liječenja CF.
Riječ iz vrlo dobrog
Iako su pregledi novorođenčadi dramatično povećali stopu dijagnoze CF kod beba, preko 25 posto dijagnoza postavlja se samo tijekom djetinjstva, tinejdžera i ranih odraslih godina.
To je problematično jer rana dijagnoza i liječenje mogu zaustaviti mnoge teže komplikacije CF-a prije nego što se bilo kakva ozbiljna šteta može učiniti. Iako liječenje ne može zaustaviti ili preokrenuti bolest, može osigurati mnogo više godina bez bolesti.
U tu svrhu važno je znati rane simptome CF i razgovarati sa svojim liječnikom ako sumnjate da vaše dijete može imati bolest. To je osobito istinito u državama koje vrše samo IRT krvne pretrage, što bi moglo rezultirati kod čak 5 posto djece ili s odgođenom dijagnozom ili s lažno negativnim rezultatom, pokazalo je istraživanje Medicinskog i javnog zdravstva Sveučilišta Wisconsin. .
Koje simptome možete očekivati kod cistične fibroze?